Sprache auswählen


angerhoefer consulting
Fachartikel
Verpackungsfehler, immer wieder Verpackungsfehler!
Artikel erschienen im März 2019 in der pharmind
Prof. Dr. Martin Angerhöfer und Dipl. Ing. Roland Kleissendorf
Verpackungsfehler sind nach wie vor der größte Anteil an Qualitätsmängeln bei Arzneimitteln. In den vergangenen Jahren betrug dieser in Deutschland stets über 40 %, ein Wert der nach Ansicht der Autoren unnötig hoch ist und mit geeigneten Maßnahmen vermeidbar wäre. Anhand ihrer Erfahrungen in der Beratungspraxis zeigen die Autoren exemplarisch an den Verpackungstypen Siegelrandbeutel, Blister mit Aluminiumdurchdrückfolie und Tablettenglasverschluss aus Kunststoff, wie Verpackungsfehler erkannt, gefunden und behoben werden können. Ebenso wichtig wie das Erkennen von Verpackungsfehlern ist die Anwendung geeigneter Vermeidungsstrategien, die ebenfalls erläutert werden. Voraussetzung für deren Einsatz ist die Kenntnis des Verpackungsvorgangs und dessen Grenzen sowie ein umfassendes Wissen der relevanten Packstoff- und Packmitteleigenschaften. Dabei sind einwandfreie, regelmäßig überprüfte und ständig angepasste Packmittelspezifikation/- beschreibungen die wesentlichen Erfolgsgaranten.
Einleitung
Wenn ein Apotheker bei einem Arzneimittel Qualitätsmängel, Nebenwirkungen oder einen falschen Gebrauch (z. B. Missbrauch) vermutet, meldet er dies gemäß den Bestimmungen der Apothekenbetriebsordnung der zuständigen Behörde und gemäß der Verpflichtung der Berufsordnungen der Apotheker an die Arzneimittelkommission der Deutschen Apotheker (AMK). Die AMK sammelt, bewertet und berichtet als Pharmakovigilanzzentrum über diese Meldungen. Zusätzlich informiert die AMK die Apotheken regelmäßig und zeitnah über neu auftretende Risiken und regulatorische Maßnahmen bei bestimmten Arzneimitteln.
Im Jahr 2018 erhielt die Geschäftsstelle der AMK insgesamt 9 486 Spontanberichte zu Qualitätsmängeln und unerwünschten Wirkungen aus 4 846 verschiedenen Apotheken. Von den 6 527 Meldungen zu Qualitätsmängeln wurden am häufigsten Verpackungsfehler gemeldet, gefolgt von galenischen Mängeln, mechanischen Defekten und Deklarationsfehlern.
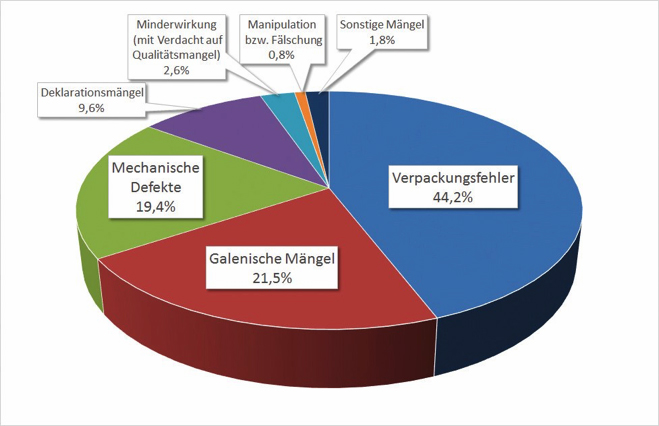
Abb. 1: Verdachtsfälle von Qualitätsmängeln bei Arzneimitteln 2018 (Quelle der Abbildung: Prof. Dr.- Ing. Martin Angerhöfer, basierend auf Daten der AMK)
Auch in den Jahren zuvor waren Verpackungsfehler der häufigste Grund für Qualitätsmängel, die der AMK gemeldet wurden.
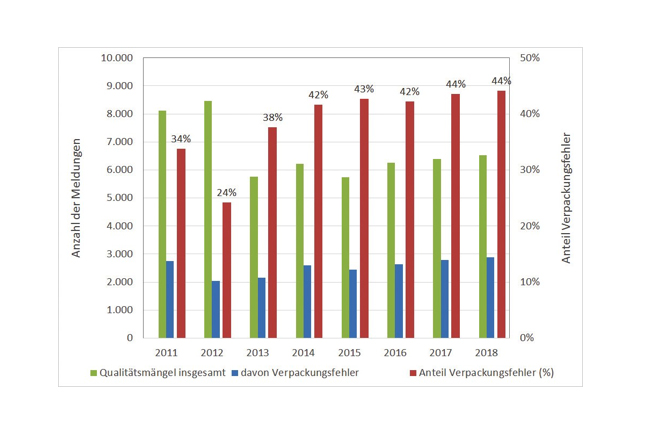
Abb. 2: Qualitätsmängel und Anteil der Verpackungsfehler 2011–2018 (Quelle der Abbildung: Prof. Dr.- Ing. Martin Angerhöfer, basierend auf Daten der AMK)
Die Autoren gehen davon aus, dass der reale Anteil von Verpackungsfehlern weit größer ist, da nicht als Verpackungsfehler direkt erkennbare Mängel die Ursache für Fehler sein können, die der Kategorie „Galenische Mängel“ zugerechnet werden. Auf ein Beispiel, in dem undichte Primärverpackungen zu Verfärbungen des Arzneimittels führten, wird später noch detaillierter eingegangen. Auch ein gewisser Anteil an Deklarationsmängeln steht in erster Linie mit Verpackungsfehlern in Verbindung.
Somit sind weit mehr als die Hälfte aller Meldungen aus dem Markt auf Fehler im Zusammenhang mit Packmitteln oder dem generellen Verpackungsvorgang zurückzuführen. Ebenso sind jeweils ein zweistelliger Prozentsatz der Rote-Hand-Briefe sowie Rückrufe des BfArM/PEI in Verbindung mit Packmittel-/Verpackungsfehlern zu sehen.
Die in diesem Beitrag verwendeten Beispiele aus der Beratungspraxis sollen die besondere Problematik der Verpackung pharmazeutischer Produkte verdeutlichen und nicht den jeweiligen Einzelfall bis ins letzte Detail erklären. Es werden keine Produktbezeichnungen und Firmennamen genannt, da diese Beispiele vollkommen unabhängig hiervon bereits jetzt oder in Zukunft wieder vorkommen können. Da die jeweiligen Verpackungstypen von vielen unterschiedlichen Firmen eingesetzt werden, sollen die Beispiele auf mögliche Schwachstellen aufmerksam machen und Wiederholungsfehler somit vermieden werden.
Beispiel 1: Siegelrandbeutel
Das erste Beispiel zeigt den Verpackungstyp „mehrlagiger Vierrand-Aluminiumverbundfolien-Beutel“, wie er für Pulver, Tabletten und Brausetabletten in großem Umfang eingesetzt wird.
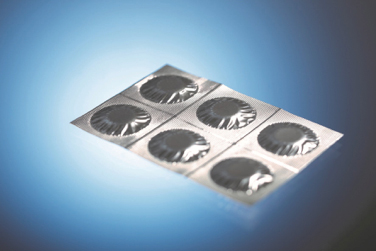
Abb. 3: Beispiel für einen Siegelrandbeutel (Quelle der Abbildung: Romaco Group, Karlsruhe)
Der meist verwendete Folienaufbau ist von innen nach außen: HD-PE als Siegelmedium, Aluminium-Folie als Barriereschicht und Papierfolie zur Bedruckung. Diese Verpackungsform wird auch außerhalb der pharmazeutischen Industrie meist auf Hochleistungsmaschinen in großen Stückzahlen seit vielen Jahrzehnten gefertigt. Bei der langjährigen Erfahrung mit diesem Packmitteltyp sollten Probleme eigentlich ausgeschlossen sein. Wenn aber bei der obligatorischen Blaubadprüfung auf Dichtigkeit bei unveränderter Folienspezifikation, unveränderter Maschineneinstellung und unveränderter Umgebung undichte Packungen gefunden werden, ist die Fehlersuche dringend geboten. Die Ursache der Undichtigkeiten waren in diesem Fall mikroskopisch kleine Risse in der Nähe des Siegelrands in Maschinenlaufrichtung, die nur punktuell und nicht eindeutig zuordenbar auftraten. Ein erster Ansatz in solchen Fällen ist immer die Fehlerzuordnung über den Rollenspiegel, um den Fehler auf bestimmte Produktionspartien und Rollen eingrenzen zu können. Ebenso erfolgt die Überprüfung der Rückhaltemuster sowie der Verbundfolien und der Einzelbestandteile des Verbunds. Werden beim erstellten Rollenspiegel erhebliche Mängel erkannt und liegen keine qualitativ nutzbaren Rückhaltemuster der eingesetzten Rollen (Aluminium und Papier) vor, wird die Fehlersuche und Fehlerzuordnung extrem schwierig. Der Materialverbund für diesen Packmitteltyp wird in einer Fertigungsanlage aus Aluminium- und Papierrollen (unterschiedlicher Herstellungspartien) mittels Extrusion (PE-Schicht) gebildet. Mithilfe einer weiteren Extrusionseinheit wird eine weitere Kunststoffschicht aufgebracht. Es entsteht fortlaufend eine Verbundfolienrolle, die am Ende des Prozesses auf mehrere Mutterrollen gewickelt und geschnitten wird. Im nächsten Arbeitsgang wird der Folienverbund auf einer anderen Maschine mit verschiedenen Aufmachungen auch für unterschiedliche Kunden bedruckt und danach in Teilrollen nach Kundenauftrag geschnitten. Durch diesen Fertigungsprozess entsteht eine ohne exakten Rollenspiegel nicht mehr zuordenbare Produktions- und Liefergrundlage. Bei dieser Ausgangslage kann die Fehlersuche nur noch mittels der gefertigten Verbundfolienpartien erfolgen, um dabei Abweichungen von der Spezifikation bzw. den früher ohne Probleme gefertigten Verbundfolien zu finden. Die sehr aufwendige Fehler-/Abweichungssuche war in diesem Fall erfolgreich. Der Fehler lag in der nicht ausreichend geglühten Aluminiumfolienpartie, die im Verbund aufgrund ihrer nicht ausreichenden Dehnung zur gefundenen Rissbildung führte.
Welche allgemein gültigen Rückschlüsse können aus diesem Fall gezogen werden? Auch langjährige Geschäftsbeziehungen bedürfen der fortlaufenden, intensiven Überprüfung durch Qualitätsaudits. Alle auf Rollen gelieferten Materialien müssen anhand eines Rollenspiegels eindeutig und sicher zuordenbar sein. Zur Überprüfung bietet sich an, eine bereits gelieferte Rolle aus dem Lagerbestand als „mangelhaft“ einzustufen und gemeinsam mit dem Lieferanten zu versuchen, die eingesetzten Halbfertigrollen bis zur Mutterrolle zu finden. Für die in der Mutterrolle eingesetzten Vormaterialien müssen Rückhaltemuster in ausreichender Menge und Qualität gelagert sein, um diese für Nachprüfungen nutzen zu können. Dies gilt natürlich auch für alle Zwischenstufen. Bereits viele Jahre genutzte Materialspezifikationen müssen in einem festgelegten Zeitrhythmus (z. B. 3 Jahre) auch ohne gemeldete Veränderung überprüft und ggf. erneuert werden. Hierzu zählen natürlich auch alle Spezifikationen der Vorlieferanten. Lieferantenwechsel und Spezifikationsänderungen sind immer dem pharmazeutischen Kunden vorab anzuzeigen und von ihm zu genehmigen. Hierzu zählen Nomenklaturänderungen, selbst wenn sie nur durch IT-Veränderungen verursacht werden. Wenn in Spezifikationen Datenblätter und Normen aufgeführt werden, muss dies immer mit Nennung des Erstellungs-/Ausgabedatums verbunden sein, da sich in diesen Unterlagen extrem wichtige Vorgaben ändern können, die z. B. beim Lieferantenwechsel erhebliche Fehlinterpretationen hervorrufen können. Abweichungen im üblichen Herstellungsprozess sind in den Lieferunterlagen zu dokumentieren und ggf. vorab mit dem Kunden zu besprechen. Alle hier aufgezählten Vorgaben müssen eindeutig und verbindlich spezifiziert werden. Sie bilden mit den übrigen Bestellunterlagen die rechtliche Grundlage der Verpackungsmateriallieferung.
Bei dem letzten Beispiel muss, abhängig vom zu verpackenden Produkt, geprüft werden, ob der Folienverbund gedehnt wird. Falls ja, muss die Dehnung mit einer ausreichenden Sicherheit spezifiziert werden. Die als Inprozesskontrolle durchgeführte Vakuumprüfung mittels Blaubad zeigt ausschließlich Undichtigkeiten, die für Flüssigkeiten durchgängig sind! Im gerade beschriebenen Fall war der Fehlerumfang wesentlich höher, da auch Beutel mit intakter und flüssigkeitsdichter Kunststofffolie, aber gerissener Aluminiumfolie gefertigt wurden.
Beispiel 2: Blister mit Aluminiumdeckfolie
Dass die als Inprozesskontrolle genutzte Blaubadprüfung nur bedingt geeignet ist, fehlerhafte Packungen zu finden, zeigt das zweite Beispiel. Die wohl häufigste Verpackungsform in der pharmazeutischen Industrie dürfte die sog. Blisterverpackung für feste Arzneiformen sein. Hier wird eine flache Folie aus Kunststoff (PVC, PVDC, PP, PET oder Kunststoffverbund) oder Aluminium auf einer Verpackungsanlage verformt, befüllt und mittels einer anderen flachen Folie, meist Aluminium (Mono-PP-Folie in sehr geringem Umfang), verschlossen. Im Normalfall bestimmt die jeweilige Gasdurchlässigkeit des Kunststoffmaterials im verformten Zustand die Dichtigkeit der Verpackung und damit die Laufzeit des verpackten Produkts – eine seit Jahrzehnten genutzte Technik, die ohne Probleme funktionieren sollte. Allerdings ergibt sich ein großes Problem, wenn bei einer 6 Monate nach dem Verpacken durchgeführten Haltbarkeitsüberprüfung feuchtigkeitsempfindlicher Tabletten mehrere Partien komplett außerhalb der Spezifikation liegen und die Überprüfung der Blister keine Anhaltspunkte auf Mängel zeigt. Auch in diesem Fall ist über die gesamten Partien hinweg alles gleich geblieben, aber die Ergebnisse unterscheiden sich. Die erste Maßnahme war, den Fertigungsablauf in die chronologische Reihenfolge zu bringen und Unterbrechungszeiten mittels Logbucheintragungen an der Verpackungsanlage herauszufinden. Hierbei zeigte sich, dass die fehlerhaften Partien alle zeitlich hintereinander gefertigt wurden. So konnte der Fehlerumfang auf wenige Partien eingeschränkt werden. Bei der Detailanalyse fiel bereits bei einer lichtmikroskopischen Untersuchung unter einer starken Vergrößerung auf, dass die Siegelstruktur nicht einheitlich und zwischen „Gut/Schlecht“- Partien unterschiedlich war. Nach einer an der Hochschule München entwickelten Methode zur Trennung von Form- und Deckfolie konnten über einem Leuchttisch die über den gesamten Blister gleichmäßig verteilten Durchrisse sichtbar gemacht werden.

Abb. 4: Im Durchlicht erkennbare Durchrisse der Aluminiumfolie (Quelle der Abbildung: Prof. Dr.-Ing. Martin Angerhöfer)
Noch besser sind die Bruchstellen der Aluminiumfolie durch einen Mikrotomschnitt quer durch den Blister zu erkennen.

Abb. 5: REM-Aufnahme mit Durchrissen der Aluminiumfolie (rote Markierung) (Quelle der Abbildung: Dipl.-Ing. Roland Kleissendorf)
In Abb. 4 ist die gelöste Aluminiumfolie im Durchlicht zu sehen. An den schwarzen Stellen ist die Folie unversehrt. Die hellen Stellen markieren die Foliendurchbrüche, die in diesem Beispiel die komplette Siegelfläche betreffen. Die Barrierefunktion der Aluminiumfolie ist damit weitgehend aufgehoben!
In Abb. 5 sind die Durchbrüche im Querschnitt gut erkennbar. Da es sich um einen Aluminium-Tiefziehblister handelt, ist die untere, unversehrte Schicht (hellgrau) die Aluminium-Formfolie. Über relativ kurze Zeit gelangt Wasserdampf durch das Siegelmedium direkt zu jeder verpackten Tablette.
Ursache des Fehlers war eine nicht korrekt eingebaute Siegelplatte. Durch technische Nachbesserungen an der Siegelstation konnte dies für die Zukunft abgestellt werden. Als weitere Absicherung wurden die Blister einer Sichtprüfung unter einer hochauflösenden Lupe unterzogen. Die Mitarbeiter in der Produktion und der Qualitätskontrolle wurden entsprechend geschult und eine Prüfanweisung erstellt.
Wichtig ist, dass die Inprozess- und Freigabeprüfung von Blisterpackungen nicht allein auf einer Blaubadprüfung fußen darf. Gerade bei Verbundmaterialien (z. B. PVC/ PVDC oder anderen Verbunden aus Kunststoff oder Aluminium) sind Fehlstellen, Risse u. Ä. nicht einfach oder überhaupt nicht auffindbar und zeigen sich erst in nicht ausreichenden Haltbarkeitsergebnissen ggf. Monate oder Jahre später. Bei Mono-Materialien (PVC, PP, PET) kann durch eine Wanddickenkontrolle die gleichmäßige Verformung überprüft und somit Abweichungen erkannt werden.
Im Rahmen dieser Reklamationsbeurteilung wurde ein komplett neues Prüfverfahren entwickelt, mit dem die Siegelnapftiefe bestimmt werden kann. Mittels lichtmikroskopischer Oberflächentopografie wurden die Siegelprofile des Blisters an definierten Stellen aufgenommen und mit einem Algorithmus ausgewertet. Ergebnisse sind beispielhaft in Abb. 6 (einwandfreier Blister) und Abb. 7 (Blister mit Fehlstellen der Alufolie) dargestellt. Es sei darauf hingewiesen, dass die Achsen mit den Prägetiefen in Abb. 6 und Abb. 7 unterschiedlich skaliert sind.
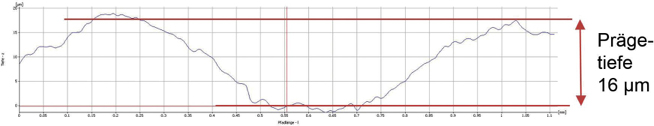
Abb. 6: Topografie und Prägetiefe eines einwandfreien Blisters (Quelle der Abbildung: Prof. Dr.-Ing. Martin Angerhöfer)
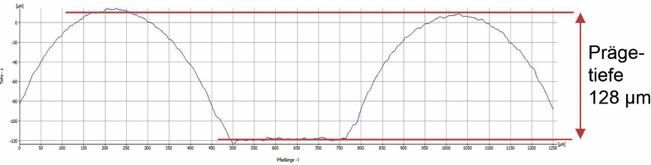
Abb. 7: Topografie und Prägetiefe eines fehlerhaften Blisters mit einer 8-fach höheren Prägetiefe als der des einwandfreien Blisters (Quelle der Abbildung: Prof. Dr.-Ing. Martin Angerhöfer)
Dieses Verfahren eignet sich in der aktuellen Form noch nicht zur Inprozesskontrolle, kann aber bei der Qualifizierung neuer Anlagen/Werkzeuge wertvolle Ergebnisse erbringen.
Beispiel 3: Tablettenglasverschluss aus Kunststoff
Ein weiterer Beratungsfall betraf einen Tablettenglasverschluss aus Kunststoff, der in unregelmäßigen Fällen nicht wie gewünscht funktionierte. Dabei bestand nach Erstöffnung die Gefahr einer Produktschädigung, da der Anwender den Verschluss nicht mehr sachgerecht verschließen konnte. In diesem Fall hatte das Unternehmen umsichtig reagiert, bevor eine Reklamation aus dem Markt auftrat. Die an der Hochschule München durchgeführten Versuche und Prüfungen zeigten, dass Klimaeinflüsse, die Handhabung und konstruktive Merkmale des Verschlusses im ungünstigen Fall zu den beobachteten Problemen führten. Da das Spritzgusswerkzeug nach jeder Produktion demontiert, in einzelne Bestandteile zerlegt und gereinigt wurde, entstand nach dem erneuten Zusammenbau immer wieder eine neue Konstellation, sodass sich jede Produktionspartie von der vorherigen unterschied. Hierdurch gab es Produktionspartien ohne Inprozessbefund, aber auch Produktionspartien mit größerer Fehlerrate. Der nun gezielt und mittels Kennzeichnung nachvollziehbar vorgenommene Zusammenbau des Werkzeugs erbrachte die gewünschte Verbesserung. In diesem Fall wurde sehr umsichtig bereits auf erste Hinweise auf ein Problem reagiert und dadurch etwaige Reklamationen vermieden.
Wichtig beim Einsatz von Werkzeugen aller Art bei Packmittellieferanten ist das Wissen, wie mit ihnen innerhalb der Packmittelfertigung umgegangen wird und welche Abweichungen hieraus resultieren können. Standzeiten von Werkzeugen dürfen nicht ausschließlich unter dem Gesichtspunkt des Lieferanten und der Kosten gesehen werden. Werkzeugverschleiß kann einschleichend zu erheblichen Funktionsproblemen führen, die nicht erkannt zu Reklamationen und bei wichtigen Funktionselementen zum Rückruf führen können. Muss dann ein Werkzeug neu erstellt und qualifiziert werden, droht ein Lieferausfall über mehrere Monate hinweg!
Fazit
Die geschilderten Beispiele zeigen u. a., dass es eine Unmenge an Detailfragen gibt, die letztendlich entscheiden, ob ein pharmazeutisches Produkt problemlos und richtig vom Verwender konsumiert bzw. angewendet werden kann. Alle sowohl für die Herstellung der Packmittel/ Packmaterialien als auch für das Fertigarzneimittel Verantwortlichen müssen jederzeit ihre internen Prozesse kennen, beobachten und auf Abweichungen konsequent reagieren. Veränderungen jeder Art müssen bekannt sein und vor Umsetzung auf mögliche Konsequenzen überdacht und überprüft werden. Das umfassende Wissen über Fertigungsmöglichkeiten und -grenzen sowie Packstoff- und Packmitteleigenschaften ist unabdingbar. Hierzu müssen Lieferant und Kunde partnerschaftlich und offen zusammenarbeiten.
Bei einigen Beratungsprojekten wurde deutlich, dass die Qualitätskontrolle inklusive der Qualitätsverantwortung an den Packmittellieferanten ausgelagert wurde und über längere Zeit hinweg beim pharmazeutischen Kunden ein Know-how-Verlust eintrat. Wenn Lieferanten dann die Qualitätskontrolle unter Kostendruck reduzieren oder gar weitgehend abbauen, ist über kurz oder lang ein Qualitätsfiasko die Folge.
Ein wesentlicher Erfolgsgarant sind einwandfreie, immer wieder überprüfte und angepasste Packmittelspezifikationen/-beschreibungen. Diese Spezifikationen müssen von den Anforderungen des Kunden her gedacht und erstellt werden sowie die für den Verpackungsprozess technisch relevanten Informationen enthalten. Zu berücksichtigen sind in gleicher Weise Vormaterialien anderer Hersteller. Die Verwendung von Datenblättern der Hersteller und allgemein gültiger Normen ist bestenfalls der Ausgangspunkt einer korrekten Spezifikationserstellung. Packmittelspezifikationen für Pri-märpackmittel sollten mindestens in einem Rhythmus von 3 Jahren komplett überprüft und überdacht werden. Um der bekannten Betriebsblindheit entgegenzuwirken, kann diese Arbeit an externe Spezialisten vergeben werden.
Sehr empfehlenswert ist es, bei der Qualifizierung neuer Verpackungsanlagen oder der Implementierung neuer Packmitteltypen im Zuge der Validierung alle Möglichkeiten der messtechnischen Prüfung der Materialien und der Packung zu nutzen. Hierdurch ergibt sich ein später nachprüfbarer Qualitätsstandard, der bei der Fehlersuche im Bedarfsfall entscheidend helfen kann, die Ursache zu finden und damit erheblich Zeit und Kosten sparen kann.
angerhoefer consulting
Prof. Dr.-Ing. Martin Angerhöfer
Eichenweg 17, 88289 Waldburg